



The US Food and Drug Administration (FDA) published its final guidance for industry on the use of real-world data (RWD) and real-world evidence (RWE) in supporting regulatory decisions for drugs and biologics on August 30, 2023.1 This guidance document represents the culmination of the draft guidance with the same title that was initially issued on December 9, 2021. According to the FDA, the guidance applies to any RWD, from registries to medical claims, and includes data on products used in clinical practice under an emergency use authorization.
This guidance represents the FDA’s current viewpoint and offers non-binding recommendations for RWD access, collection and analysis transparency, study monitoring and reporting, and the sponsor’s responsibilities. The guidance mainly centers on non-interventional clinical study designs, such as observational cohort studies and case-control studies. It also describes the FDA’s perspective on potential applications of RWD in interventional studies conducted under an investigational new drug (IND) application. For example, RWD might help identify potential participants for a randomized controlled trial, choose study endpoints, or serve as a comparison group in an externally controlled trial.
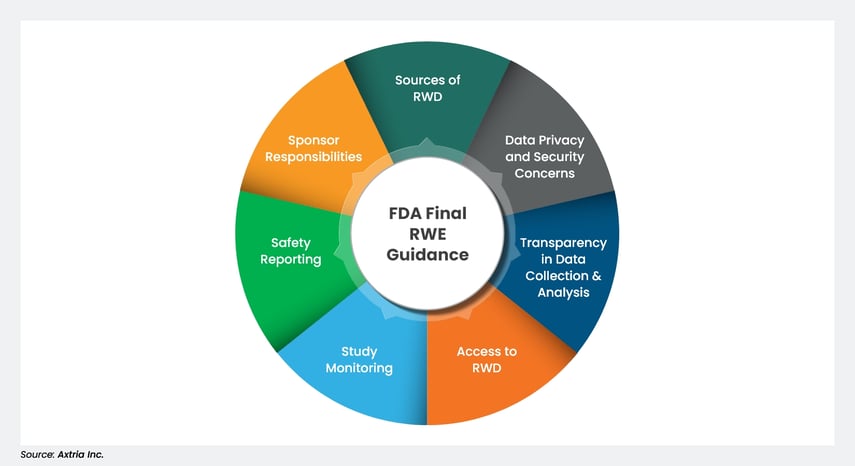
Figure 1: Topics included in the FDA’s Final Guidance on RWE/RWD.
Applicability of 21 CFR Part 312
This section of the guidance discusses the applicability of the US Code of Federal Regulations, Title 21, Part 312 (Investigational New Drug Application) to studies involving the use of RWD. Part 312 outlines the procedures and conditions governing the use of investigational new drugs, including the requirements for an IND submission to the FDA and its review.
- The FDA recognizes the potential utility of using RWD in interventional studies, for example, identifying potential participants for a randomized controlled trial to ascertain endpoints or outcomes.
Regulatory Considerations for Non-Interventional Studies
The FDA’s final guidance also lays out considerations for RWD in non-interventional studies, such as observational studies or trials in which treatments are prescribed as originally authorized.
- Sources of RWD: Various sources of RWD can be analyzed in non-interventional studies, including registries, electronic health records (EHRs), and medical claims. The topics discussed in this guidance apply to any type of RWD, including data on products used in clinical practice under an emergency use authorization.
- Data privacy and security concerns: When appropriate, sponsors should consult with experts in data privacy issues as part of the design process for a non-interventional study. These experts will help identify and address data privacy and security concerns raised when accessing healthcare data.
- Transparency regarding data collection and analysis: The FDA should be engaged early in the study design and protocol process to ensure transparency in data collection and analysis.
- Access to RWD: The FDA should be allowed access to any patient-level data used as part of the clinical study included in a marketing application.
- Study monitoring: The FDA’s final guidance defines study monitoring as “one of the principal quality control activities” in RWE generation. The agency calls on sponsors to use a “risk-based quality management approach to study oversight” to ensure the data is reliable and protected and the study is conducted according to protocol.
- Safety reporting: Non-interventional studies used to support regulatory applications are subject to the standard post-marketing safety reporting regulations regarding the occurrence of relevant adverse events.
- Sponsor responsibilities: These include detailing the governance procedures needed to ensure the appropriate design, conduct (including data analysis), and oversight of the RWE studies.
The FDA guidance offers valuable pointers for using RWE in regulatory decisions. By following this guidance and collaborating with the FDA from the inception of a study, industry professionals can better use RWE to improve drug and biological product safety and effectiveness evaluations.
Axtria’s solutions in RWE and HEOR take an integrated approach, encompassing data infrastructure, analytics and talents, processes, and governance for agile and actionable insight deliveries. Axtria offers integrated evidence generation by leveraging a full spectrum of analytics (descriptive, diagnostic, predictive, and counterfactual) across the product lifecycle, encompassing pre-launch, launch, and post-launch. Our innovative solutions bridge the evidence gap between clinical trials and real-world practice. We can help clients in navigating the ever-evolving regulatory landscape.
We hope you find this summary of the FDA’s final RWE guidance valuable. If you would like more information on Axtria’s solutions in RWE and HEOR, please contact us at connect@axtria.com.
References
- US Dept. of Health and Human Services. Considerations for the Use of Real-World Data and Real-World Evidence to Support Regulatory Decision-Making for Drug and Biological Products: Guidance for Industry. Food and Drug Administration, et al. August 30, 2023. Accessed November 7, 2023. https://www.fda.gov/media/171667/download
Author details

